



-
摘要:
在未知材料化学成分和性能关系的情况下,通过传统的“试错-纠错”方法研发具有特定功能的新材料成本高且经常失败。随着人工智能和数据驱动的第四科学范式的发展,材料基因工程(MGE)已经成为材料设计与研发的新模式。综述了材料基因工程中高通量计算、材料数据库和人工智能方法的研究进展。介绍了材料高通量计算常用的框架和方法; 阐述了材料数据库在材料数据类型和数据标准两方面的发展现状和有待解决的难题; 总结了人工智能方法在材料关键基础问题中的应用。从高通量可视化计算方法、材料多类型数据库和可视化机器学习框架三方面重点证述了自主开发的多尺度集成可视化的高通量自动计算和数据管理智能平台ALKEMIE。展望了材料基因工程未来的发展趋势。
-
关键词:
- 材料基因工程(MGE) /
- 高通量计算 /
- 材料数据库 /
- 机器学习 /
- 智算平台
Abstract:Without knowing the relationship between the chemical composition and properties of the material, the development of new materials with desired properties based on conventional trial-and-error methods is cost inefficient and sometimes ends fruitlessly. With the development of artificial intelligence and the fourth science paradigm, data-driven science, materials genetic engineering (MGE) has become a new approach for designing novel materials. In this paper, recent advances in high-throughput computation, materials database, and artificial intelligence methods in MGE are reviewed. First, the framework and software for high-throughput computations of materials are introduced. Then, the progress and critical problems of materials databases are presented in terms of types and standard interfaces of materials data. Next, we summarize the applications of artificial intelligence methods in critical issues of materials science. In particular, from the aspects of visualized high-throughput calculation methods, multi-type materials database and visualized machine learning framework, we emphasize an in-house developed platform ALKMIE, featured by multi-scale integration of automatic high-throughput calculation, visualization, and intelligent data management. Finally, we highlight the future directions of MGE.
-




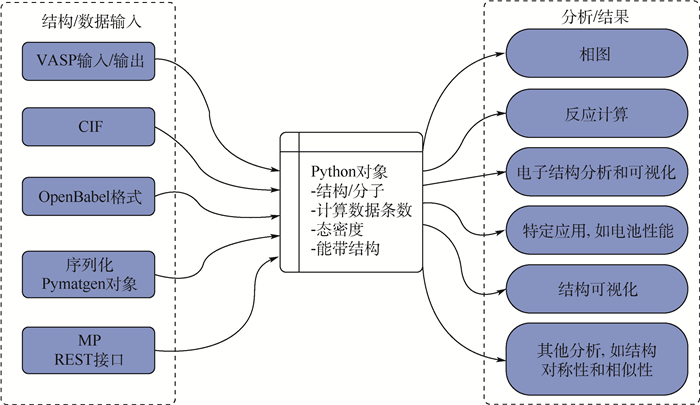
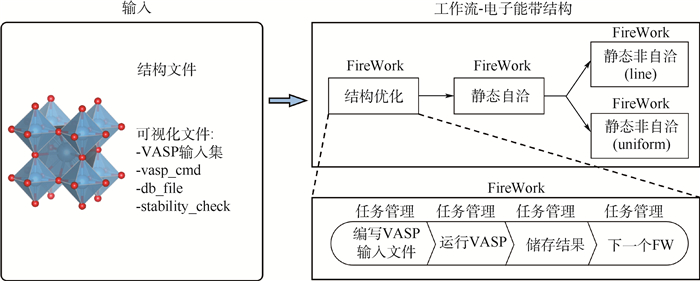
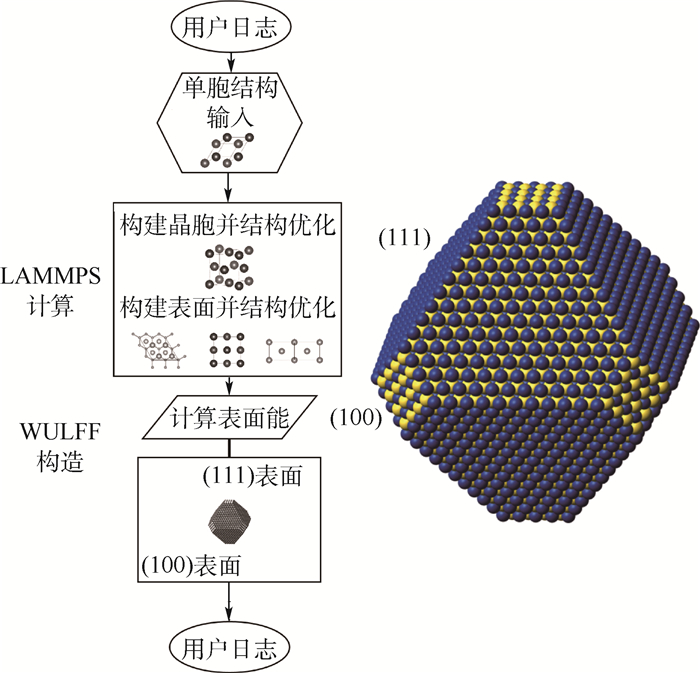
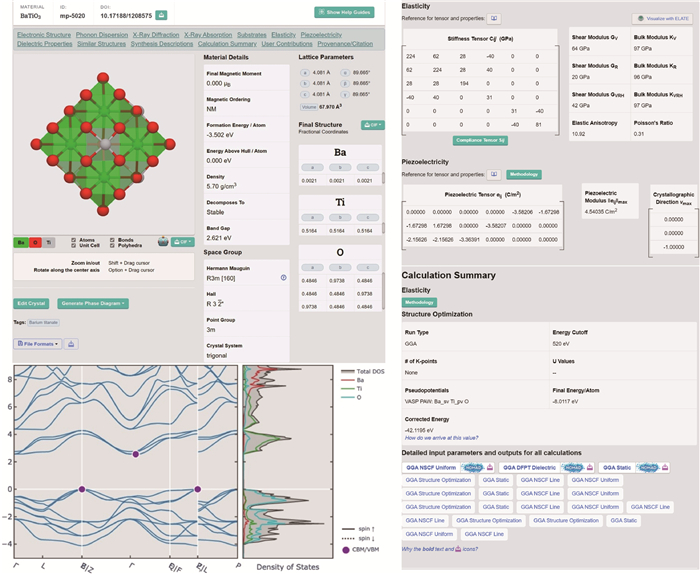
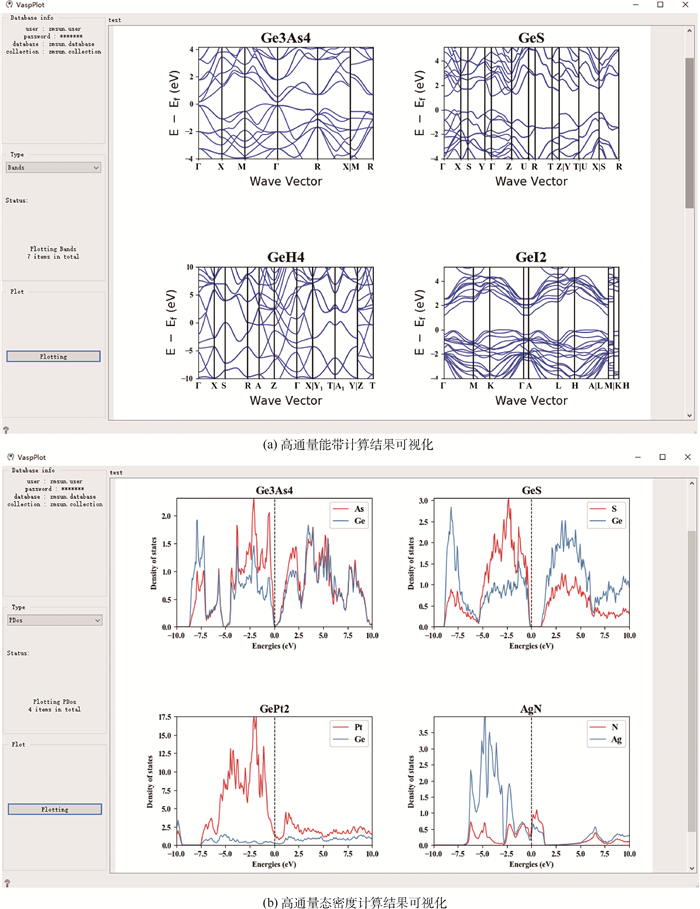
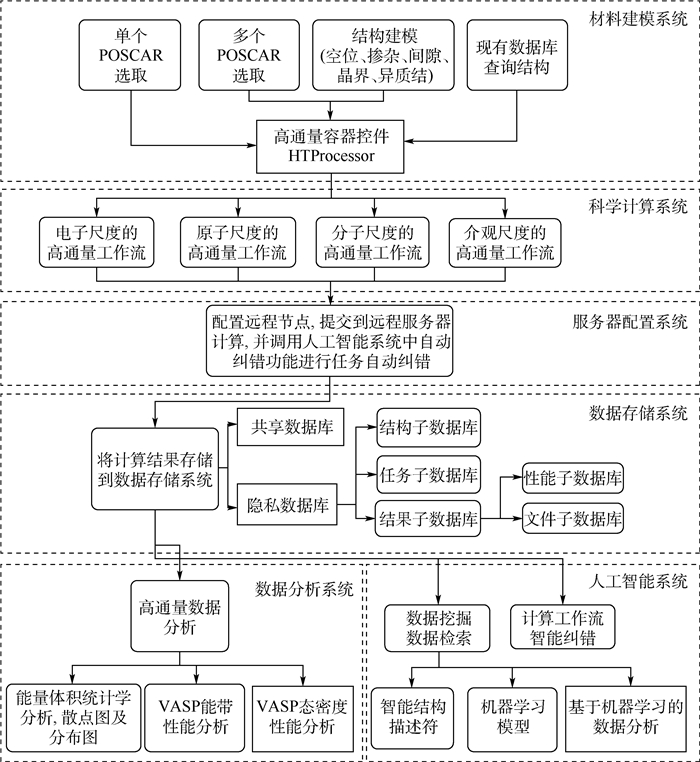
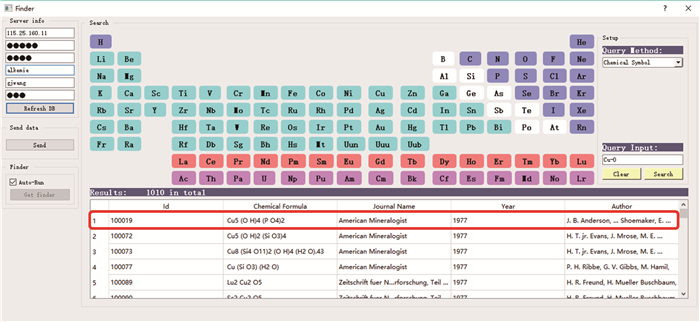
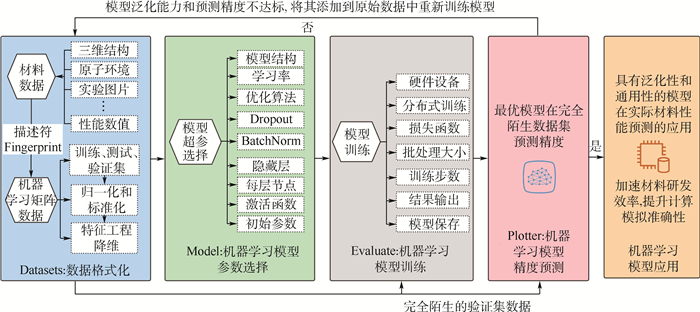
图 8 多尺度集成可视化的高通量自动计算和数据管理智能平台ALKEMIE计算模块概况
Figure 8. Overview of platform with multi-scale integration of visualized automatic high-throughput calculation and intelligent data management ALKEMIE
表 1 材料高通计算软件和框架发展现状
Table 1. Software and frameworks of material high-throughput calculation
名称 说明 网址 ALKEMIE 高通量计算框架和软件 https://alkemine.org/ Pymatgen 高通量计算框架 http://pymatgen.org/ FireWorks 高通量计算框架 https://materialsproject.github.io/fireworks/ Atomate 高通量计算框架和软件 https://atomate.org/ AFLOW-π 高通量计算软件 http://aflowlib.org/scr/aflowpi/ ASE 高通量计算框架和软件 http://wiki.fysik.dtu.dk/ase/ AiiDA 高通量计算框架和软件 http://aiida.net/ Abipy 高通量计算软件 https://github.com/abinit/abipy MPInterfaces 高通量计算框架 http://henniggroup.github.io/MPInterfaces/ Imeall 高通量计算软件 https://github.com/Montmorency/imeall MedeA 高通量计算软件 https://www.materialsdesign.com/ IprPy 高通量计算框架 https://www.ctcms.nist.gov/potentials/iprPy/ Pylada 高通量计算框架 http://pylada.github.io/pylada MIP 高通量计算软件 http://www.mip3d.org/ MatCloud 高通量计算软件 http://matcloud.cnic.cn/ JAMIP 高通量计算框架 http://jamip-code.com MatAi 高通量计算框架 https://www.mat.ai/ 中国材料基因工程高通量计算平台 高通量计算框架 http://mathtc.nscc-tj.cn/  下载: 导出CSV
下载: 导出CSV
表 2 材料多类型数据库的发展
Table 2. Development of multi-type databases of materials
下载: 导出CSV
-
[1] XIANG X, SUN X, BRICENO G, et al. A combinatorial approach to materials discovery[J]. Science, 1995, 268(5218): 1738-1740. doi: 10.1126/science.268.5218.1738 [2] ALLISON J. Integrated computational materials engineering: A perspective on progress and future steps[J]. JOM, 2011, 63(4): 15-18. doi: 10.1007/s11837-011-0053-y [3] LIU Z. High throughput CALPHAD modeling and the materials genome[J]. Technology, 2010, 617: 622. [4] LIU Z. View and comments on the dataecosystem: Ocean of data[J]. Engineering, 2020, 6(6): 604-608. doi: 10.1016/j.eng.2020.04.009 [5] WHITE A. The materials genome initiative: One year on[J]. MRS Bulletin, 2012, 37(8): 715-716. doi: 10.1557/mrs.2012.194 [6] AGRAWAL A, CHOUDHARY A. Perspective: Materials informatics and big data: Realization of the "fourth paradigm" of science in materials science[J]. APL Materials, 2016, 4(5): 053208. doi: 10.1063/1.4946894 [7] JAIN A, ONG S, HAUTIER G, et al. Commentary: The materials project: A materials genome approach to accelerating materials innovation[J]. APL Materials, 2013, 1(1): 011002. doi: 10.1063/1.4812323 [8] CURTAROLO S, SETYAWAN W, HART G, et al. AFLOW: An automatic framework for high-throughput materials discovery[J]. Computational Materials Science, 2012, 58: 218-226. doi: 10.1016/j.commatsci.2012.02.005 [9] LARSEN A, MORTENSEN J, BLOMQVIST J, et al. The atomic simulation environment-A Python library for working with atoms[J]. Journal of Physics: Condensed Matter, 2017, 29(27): 273002. doi: 10.1088/1361-648X/aa680e [10] PIZZI G, CEPELLOTTI A, SABATINI R, et al. AiiDA: Automated interactive infrastructure and database for computational science[J]. Computational Materials Science, 2016, 111: 218-230. doi: 10.1016/j.commatsci.2015.09.013 [11] GONZE X, AMADON B, ANGLADE P M, et al. ABINIT: First-principles approach to material and nanosystem properties[J]. Computer Physics Communications, 2009, 180(12): 2582-2615. doi: 10.1016/j.cpc.2009.07.007 [12] MATHEW K, SINGH A K, GABRIEL J J, et al. MPInterfaces: A materials project based Python tool for high-throughput computational screening of interfacial systems[J]. Computational Materials Science, 2016, 122: 183-190. doi: 10.1016/j.commatsci.2016.05.020 [13] LAMBERT H, FEKETE A, KERMODE J R, et al. Imeall: A computational framework for the calculation of the atomistic properties of grain boundaries[J]. Computer Physics Communications, 2018, 232: 256-263. doi: 10.1016/j.cpc.2018.04.029 [14] WANG G J, PENG L, LI K, et al. ALKEMIE: An intelligent computational platform for accelerating materials discovery and design[J]. Computational Materials Science, 2021, 186: 110064. doi: 10.1016/j.commatsci.2020.110064 [15] WANG G J, LI K, PENG L, et al. High-throughput automatic integrated material calculations and data management intelligent platform and the application in novel alloys[J]. Acta Metallurgica Sinica, 2021, 58(1): 75-88. [16] YANG J, LI H, WU T, et al. Evaluation of half-Heusler compounds as thermoelectric materials based on the calculated electrical transport properties[J]. Advanced Functional Materials, 2008, 18(19): 2880-2888. doi: 10.1002/adfm.200701369 [17] YANG X, WANG Z, ZHAO X, et al. MatCloud: A high-throughput computational infrastructure for integrated management of materials simulation, data and resources[J]. Computational Materials Science, 2018, 146: 319-333. doi: 10.1016/j.commatsci.2018.01.039 [18] ZHAO X G, ZHOU K, XING B, et al. JAMIP: An artificial-intelligence aided data-driven infrastructure for computational materials informatics[J]. Science Bulletin, 2021, 66(19): 1973-1985. doi: 10.1016/j.scib.2021.06.011 [19] CURTAROLO S, SETYAWAN W, WANG S, et al. AFLOWLIB. ORG: A distributed materials properties repository from high-throughput ab initio calculations[J]. Computational Materials Science, 2012, 58: 227-235. doi: 10.1016/j.commatsci.2012.02.002 [20] CURTAROLO S, HART G L, NARDELLI M B, et al. The high-throughput highway to computational materials design[J]. Nature Materials, 2013, 12(3): 191-201. doi: 10.1038/nmat3568 [21] HU S, LIU B, LI Z, et al. Identifying optimal dopants for Sb2Te3 phase-change material by high-throughput ab initio calculations with experiments[J]. Computational Materials Science, 2019, 165: 51-58. doi: 10.1016/j.commatsci.2019.04.028 [22] GAN Y, MIAO N, LAN P, et al. Robust design of high-performance optoelectronic chalcogenide crystals from high-throughput computation[J]. Journal of the American Chemical Society, 2022, 144(13): 5878-5886. doi: 10.1021/jacs.1c12620 [23] BELSKY A, HELLENBRANDT M, KAREN V L, et al. New developments in the inorganic crystal structure database (ICSD): Accessibility in support of materials research and design[J]. Acta Crystallographica, 2002, 58(3): 364-369. doi: 10.1107/S0108768102006948 [24] QUIRÓS M, GRAŽULIS S, GIRDZIJAUSKAITÉ S, et al. Using SMILES strings for the description of chemical connectivity in the crystallography open database[J]. Journal of Cheminformatics, 2018, 10(1): 1-17. doi: 10.1186/s13321-017-0256-5 [25] SAAL J E, KIRKLIN S, AYKOL M, et al. Materials design and discovery with high-throughput density functional theory: The open quantum materials database (OQMD)[J]. JOM, 2013, 65(11): 1501-1509. doi: 10.1007/s11837-013-0755-4 [26] DRAXL C, SCHEFFLER M. The NOMAD laboratory: From data sharing to artificial intelligence[J]. Journal of Physics: Materials, 2019, 2(3): 036001. doi: 10.1088/2515-7639/ab13bb [27] OGATA T, YAMAZAKI M. New stage of MatNavi, materials database at NIMS[C]//ASME Pressure Vessels and Piping Conference. New York: ASME, 2009: 1561-1568. [28] CHOUDHARY K, DECOST B, TAVAZZA F. Machine learning with force-field-inspired descriptors for materials: Fast screening and mapping energy landscape[J]. Physical Review Materials, 2018, 2(8): 083801. doi: 10.1103/PhysRevMaterials.2.083801 [29] BORYSOV S S, GEILHUFE R M, BALATSKY A V. Organic materials database: An open-access online database for data mining[J]. PLOS One, 2017, 12(2): 171501. [30] HUMMELSHØJ J S, ABILD-PEDERSEN F, STUDT F, et al. CatApp: A Web application for surface chemistry and heterogeneous catalysis[J]. Angewandte Chemie International Edition, 2012, 51(1): 272-274. doi: 10.1002/anie.201107947 [31] ANDERSEN C W, ARMIENTO R, BLOKHIN E, et al. OPTIMADE, an API for exchanging materials data[J]. Scientific Data, 2021, 8(1): 217. doi: 10.1038/s41597-021-00974-z [32] LIN X, YANG W, WANG K L, et al. Two-dimensional spintronics for low-power electronics[J]. Nature Electronics, 2019, 2(7): 274-283. doi: 10.1038/s41928-019-0273-7 [33] LU S, ZHOU Q, OUYANG Y, et al. Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning[J]. Nature Communications, 2018, 9(1): 3405. doi: 10.1038/s41467-018-05761-w [34] WU Y, LU S, JU M G, et al. Accelerated design of promising mixed lead-free double halide organic-inorganic perovskites for photovoltaics using machine learning[J]. Nanoscale, 2021, 13(28): 12250-12259. doi: 10.1039/D1NR01117K [35] ZHANG H, FU H, ZHU S, et al. Machine learning assisted composition effective design for precipitation strengthened copper alloys[J]. Acta Materialia, 2021, 215: 117118. doi: 10.1016/j.actamat.2021.117118 [36] VILLARS P, CENZUAL K, GLADYSHEVSKⅡ R, et al. PAULING FILE-towards a holistic view[J]. Chemistry of Metals and Alloys, 2018, 11(3): 43-76. [37] TSHITOYAN V, DAGDELEN J, WESTON L, et al. Unsupervised word embeddings capture latent knowledge from materials science literature[J]. Nature, 2019, 571(7763): 95-98. doi: 10.1038/s41586-019-1335-8 [38] KONONOVA O, HUO H, HE T, et al. Text-mined dataset of inorganic materials synthesis recipes[J]. Scientific Data, 2019, 6(1): 203. doi: 10.1038/s41597-019-0224-1 [39] BLANK T B, BROWN S D, CALHOUN A W, et al. Neural network models of potential energy surfaces[J]. The Journal of Chemical Physics, 1995, 103(10): 4129-4137. doi: 10.1063/1.469597 [40] MALSHE M, NARULKAR R, RAFF L M, et al. Development of generalized potential-energy surfaces using many-body expansions, neural networks, and moiety energy approximations[J]. The Journal of Chemical Physics, 2009, 130(18): 184102. doi: 10.1063/1.3124802 [41] BEHLER J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials[J]. The Journal of Chemical Physics, 2011, 134(7): 074106. doi: 10.1063/1.3553717 [42] GASTEGGER M, SCHWIEDRZIK L, BITTERMANN M, et al. wACSF: Weighted atom-centered symmetry functions as descriptors in machine learning potentials[J]. The Journal of Chemical Physics, 2018, 148(24): 241709. doi: 10.1063/1.5019667 [43] YAO K, HERR J E, TOTH D W, et al. The TensorMol-0.1 model chemistry: A neural network augmented with long-range physics[J]. Chemical Science, 2018, 9(8): 2261-2269. doi: 10.1039/C7SC04934J [44] SOSSO G C, MICELI G, CARAVATI S, et al. Neural network interatomic potential for the phase change material GeTe[J]. Physical Review B, 2012, 85(17): 174103. doi: 10.1103/PhysRevB.85.174103 [45] ARTRITH N, URBAN A. An implementation of artificial neural-network potentials for atomistic materials simulations: Performance for TiO2[J]. Computational Materials Science, 2016, 114: 135-150. doi: 10.1016/j.commatsci.2015.11.047 [46] MOCANU F, KONSTANTINOU K, LEE T, et al. Modeling the phase-change memory material, Ge2Sb2Te5, with a machine-learned interatomic potential[J]. The Journal of Physical Chemistry B, 2018, 122(38): 8998-9006. doi: 10.1021/acs.jpcb.8b06476 [47] ZHANG L, HAN J, WANG H, et al. Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics[J]. Physical Review Letters, 2018, 120(14): 143001. doi: 10.1103/PhysRevLett.120.143001 [48] CHEN Q, CHEN M, ZHU L, et al. Composition gradient mediated semiconductor metal transition in ternary transition metal dichalcogenide bilayers[J]. ACS Applied Materials & Interfaces, 2020, 12(40): 45184-45191. [49] PENG Q, ZHOU J, CHEN J, et al. Cu single atoms on Ti2CO2 as a highly efficient oxygen reduction catalyst in a proton exchange membrane fuel cell[J]. Journal of Materials Chemistry A, 2019, 7(45): 26062-26070. doi: 10.1039/C9TA08297B [50] GAN Y, WANG G, ZHOU J, et al. Prediction of thermoelectric performance for layered IV-V-VI semiconductors by high-throughput ab initio calculations and machine learning[J]. NPJ Computational Materials, 2021, 7(1): 176. doi: 10.1038/s41524-021-00645-y -
 下载:
下载:

 点击查看大图
点击查看大图

计量
- 文章访问数: 729
- HTML全文浏览量: 188
- PDF下载量: 115
- 被引次数: 0
